The complete guide to SaMD (software as a medical device)
Software has 'eaten' many industries ever since investor Marc Andreesen penned his famous column thirteen years ago. And medical device software is no exception.
Software is transforming the way technicians detect diseases, how doctors suggest treatments, and how patients manage their health. Far from acting as a support or being tied to a single medical device, software itself is now a discrete product, known as software as a medical device (SaMD).
SaMD provides more effective ways of delivering healthcare at scale. And regulatory premarket submission guidelines give clear guidance on how companies should plan, develop and deploy their SaMD and software in a medical device (SiMD) products.
When you have a firm grasp of the various nuances of SaMD solutions and the regulations that govern SaMD development, it becomes easier to not only navigate regulation, but also how to launch and market those products more efficiently.
What is software as a medical device (SaMD)?
SaMD definition
The International Medical Device Regulators Forum (IMDRF), which consists of medical device regulators from around the world including the FDA, defines SaMD as any software used for medical purposes that isn't part of the hardware itself.
In other words, SaMD doesn’t need to be part of another device to achieve its intended medical purpose. It can be an in vitro diagnostic (IVD) medical device, a medical mobile app or a desktop software, just to name a few examples.
And the software can be run on smartphones, tablets, laptops, or other electronic devices at most user’s fingertips.
Since SaMD doesn’t need a specific medical device to function, it can interface with various medical devices to carry out medical functions, ranging from diagnosis and prevention to the monitoring and treatment of diseases and physiological conditions.
As hardware and software increasingly intertwine in the 21st-century medical device world, it can sometimes be difficult to determine the category your device falls into.
Here's a handy guide to help you determine if your medical device should be treated as a hardware or SaMD product:

How medical device software works
SaMD takes in various data inputs, like lab results, patient symptoms, or imaging data, and processes them using various types of algorithms. The software then produces outputs that are used for various medical purposes, including for helping users in diagnosing, treating, and managing their health issues.
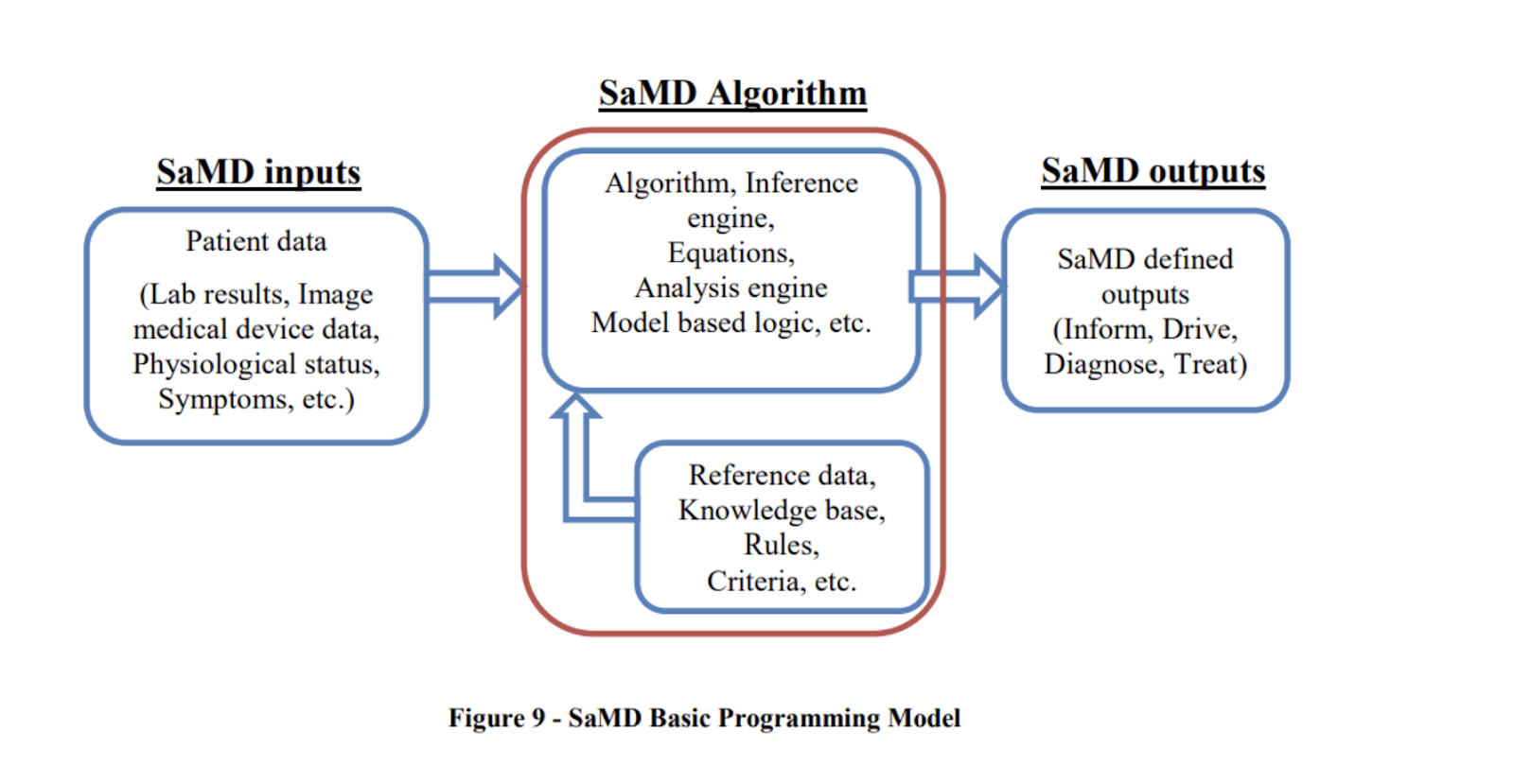
SaMD basic programming model, SOURCE: FDA
SaMD can also use sensors, smartphones and external sources to collect relevant data that’s pushed into the software algorithm.
Software as a medical device examples
The FDA has provided clarification on which products should be considered SaMD and which shouldn’t. Software developers whose products are considered SaMD must seek relevant regulatory approvals. Though this list is far from exhaustive, here are several examples of what the FDA considers SaMD products:
- Software that collects patient data to assist doctors in planning a linear accelerator cancer treatment
- Software that analyzes personalized patient data to suggest the proper drug dose
- Software that uses imaging data to track the size of a mole and decide the risk of melanoma cancer
- Software that analyzes magnetic resonance imaging (MRI) data to detect and diagnose a stroke
- Software that collects data from various digital devices to determine users’ risk factors related to epileptic seizures
Not every software product used in hospitals or medical settings is a SaMD. Here are examples of products the FDA doesn’t consider SaMD:
- Software that manages an infusion pump’s motors and pumping operations
- Software that controls an implantable pacemaker
- Software that encrypts medical device data for transmission
- Software for patient registration, video and voice calling, and scheduling visits
- Software that monitors the performance of medical devices for servicing purposes
Does SaMD require FDA approval?
Regulating SaMD has proven to be a challenge for the FDA. One reason is the agency is used to reviewing medical devices that remain unchanged by their manufacturers once on the market. A stent cleared by the FDA, for instance, is implanted into the patient and won’t be changing any of its features once placed.
But SaMD works in a different way. Software developers can make continuous changes to their products, whether it’s security updates, new features, or product evolutions. Should each of these changes first be approved by the FDA? The agency can take up to eight months to approve medical devices, depending on whether developers submit a 510(k) application, self-register, or submit a Premarket Approval (PMA) application.
The FDA is aware that delays in approving products can negatively impact patients and producers. In its Digital Health Innovation Action Plan, the FDA says lengthy approval of updates for SaMD products may prevent patients from accessing critically important technologies.
Then there’s also the issue of many SaMD developers not being experienced in dealing with the FDA. Studies show that it takes 3 to 7 years to bring a medical device to market. Inexperienced developers may struggle with getting their products approved and shipped on time, ending up discouraged and abandoning their work on SaMD products. Products that present moderate- to high-risk levels to patient health may involve an especially rigorous evaluation process.
Lastly, SaMD products can continually gather medical data, such as medical images, lab results, heart rate, body temperature, and more. As such, these products and patients’ data need to be well protected against potential cybersecurity breaches.
By the end of Q1 of 2024, for instance, over 13 patient records have been exposed in online attacks in the U.S. alone. SaMD products and development processes should therefore comply with HIPAA standards. Failure to do so may enable attackers to leak sensitive information and leave SaMD providers to deal with substantial fines and even prison sentences.
See how one of our customers got ready for their SaMD 510(k) submission in just 90 days
FDA guidance on software as a medical device
In June 2023, the FDA's 'Content of Premarket Submissions for Device Software Functions' replaced the 2005 'Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices'.
The guidance sets out a risk-based approach to the documentation level, which can be basic or enhanced.
The FDA classifies SaMD products into four categories.
Level I and Level II category products, like a stress monitoring app that provides no diagnosis, are those with the lowest impact on patient safety or public health and require basic documentation.
Developers of Level III and IV products, such as a software product that monitors and analyzes heart rate and blood pressure data in intensive care units, whose failure can lead to death or serious injury to patients or others, will need to provide enhanced documentation for their premarket submissions.
Enhanced documentation is also needed if products are intended to be used to test for transfusion-transmitted infections in blood donations or to determine donor and recipient compatibility.

Depending on their documentation level, product developers are supposed to provide various types of documents, including:
- Overview of software features
- Detailed diagrams
- Flow of data for user interaction
- Risk management plan
- Revision history
- List of unresolved bugs
The FDA has also provided an example of how applicants can clarify their software architecture in premarket submissions.
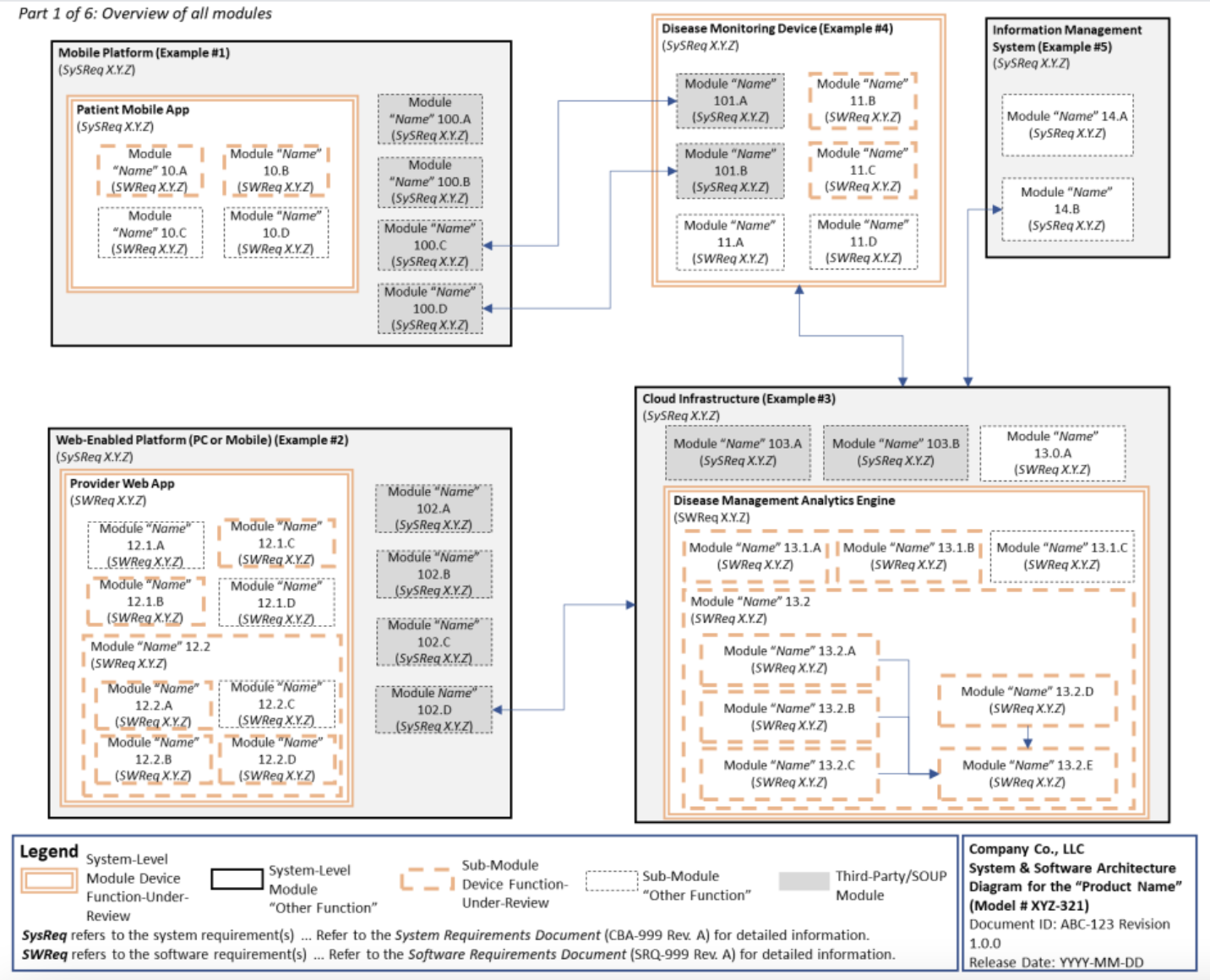
An example of a software architecture diagram chart FDA would want to see in premarket submissions, SOURCE: FDA
Benefits of software as a medical device
SaMD products offer a number of benefits to patients, physicians, and software developers. Some of these benefits are:
- Patients can play a more active role in managing their health. For instance, patients with asthma can use smart sensor data to identify and avoid triggers of flare-ups.
- Powerful SaMD tools could even outperform trained clinicians in making medical diagnoses and earlier detection of certain diseases. For example, an AI model built by researchers from Google Health and Imperial College London has proven to be more accurate than doctors in identifying breast cancer from mammograms.
- SaMD products make it easy for software developers to iterate a product and drive innovation. Developers can gather data and solicit user feedback quickly, which shortens the feedback loop and time to market.
View our inspiring list of the top medical device start-ups to keep an eye on in 2024
Challenges of software as a medical device
SaMD products are also posing various challenges to its developers, regulators, and users. Some of these challenges include:
- Developers might need more time than usual to get products built. Patient safety and regulatory considerations add new complexities to the existing product development methodologies.
- Regulators have to walk a fine line between ensuring patient safety and supporting innovation. Too many strict regulations might disincentivize companies from investing in new products, but authorities also can’t compromise patient safety.
- Cybersecurity vulnerabilities pose a risk to users and developers of SaMD products. Attackers may breach products, steal sensitive data, and affect the functionality of software.
To tackle these challenges in a controlled, holistic manner that ensures customer and regulatory requirements are met, you'll need to build a quality management system for your SaMD company.
And to help you get to grips with your quality and regulatory requirements, it might not be a bad idea to turn to one of the best medical device consultants to guide you through the process.
Artificial intelligence/machine learning-based SaMD products
Software developers also use machine learning (ML), a type of artificial intelligence (AI), to create ever more sophisticated SaMD products. ML spots patterns in data and adapts accordingly.
In theory, ML-based products become more valuable over time as they gather and analyze more data. For example, a smart sensor device that predicts the chance of its user having a heart attack becomes more accurate at predicting outcomes the more data it gathers.
Read our breakdown of the EU AI Act and what it means for SaMD companies
In April 2019, the FDA released a discussion paper that described potential approaches to premarket review for AI/ML-driven SaMD products.
The proposed framework was based on the principle of a 'predetermined change control plan': companies will have to describe aspects of a product that may be changed through algorithmic learning as well as how the algorithm is to learn while remaining safe for use.
Under this framework, the FDA would expect developers to monitor their SaMD products in real time. The agency would also require them to provide periodic updates on the changes that were implemented based on the approved specifications and protocols.
The FDA received feedback on its discussion paper from SaMD product developers, and in January 2021 unveiled its 'AI/ML SaMD Action Plan'.
The 5-part framework laid out the following steps:
- Update the proposed framework on regulating AI/ML-based SaMD products
- Encourage tech communities to harmonize Good Machine Learning Practice (GMLP) principles (more on those below!)
- Promote transparency in how companies label and market AI/ML-based SaMD products
- Support efforts to develop methods for identifying and removing algorithm bias
- Provide more clarity on how SaMD developers should monitor real-world performance of their products
The Agency then followed up the Action Plan with its 2023 'marketing submission recommendations' and, in March 2024, mapped out its coordinated AI framework in its AI & Medical Products whitepaper.
This slew of documents shows the FDA coming to terms with the rapidly spreading AI market, and now offers a much more solid and comprehensive regulatory framework for AI SaMD developers than was the case.
Good Machine Learning Practice principles for AI/ML-based SaMD products
GxP is critical in life science, and SaMD development is no different.
For AI-based SaMD, good machine learning practice (GCLP) is a critical quality element to consider.
The FDA, the UK’s Medicines and Healthcare Products Regulatory Agency (MHRA), and Health Canada have identified 10 principles that can guide the development of GMLP. These principles are:
- Products are to be developed by a multidisciplinary team
- Product teams follow good software development practices
- Study participants and datasets should be representative of the target patient population
- Product teams will select and keep training data sets independent of test sets
- Companies will use the best available methods to develop a reference dataset
- Developers design the product that supports users in achieving the intended goal
- When relevant, the focus is placed on the performance of the human-AI team
- Developers test the product in clinically relevant conditions
- Users are given clear information on the product’s intended use
- Product teams will monitor deployed products for performance and risks
While these principles aren’t an official regulation SaMD developers must follow, adhering to these FDA-endorsed best practices will make it easier to get regulatory approvals for medical products.
SaMD regulatory compliance
The future of SaMD is undeniably exciting. Software has the power to improve the diagnosis and treatment of various diseases and enable patients to better manage their own health. And the data these products gather can lead to faster innovation and improved algorithms.
But the push for innovation shouldn’t come at the expense of patient safety and clinical efficacy. Staying on top of the FDA's recently updated regulatory expectations is vital for SaMD companies.
Although it might seem a daunting task to bring your SaMD product to the world, having a sophisticated SaMD electronic quality management system (eQMS) in place makes things easier. An eQMS helps SaMD companies not only manage critical documentation, but also connects various project components like device risks, allowing you to spot and fix issues and better manage the impact of software changes and updates.
For companies developing cutting-edge medical software, turning to software tools of their own is a natural step to ensure their quality is optimized, airtight and best-in-class.
