The EU IVDR: everything you need to know (Ultimate Guide)
The In Vitro Diagnostic Regulation (IVDR) (EU) 2017/746, better known as the EU IVDR, goes live today!
After a 5-year transition period, from May 26, 2022 every business marketing in vitro diagnostic medical devices to European Union territories must meet the requirements of the regulation.
Listen to the audio version of this article read by a real person here (Sound on!):
What is the IVDR? And what does it really mean for your business? Find out with our complete guide.
Table of Contents
What is the IVDR?
Simply put: the IVDR is the EU's new guiding regulation specifying the safety, integrity and quality requirements for any medical device that performs an in vitro diagnostic function.
3-second Latin lesson
In vitro = 'in glass', referring to the capture and analysis of biological samples outside the body
An in vitro device includes any:
- Reagants and reagant products
- Calibrators
- Instruments
- Kits
- Software and systems
- Specimen receptacles
which examine and analyze bodily specimens for the purpose of providing information about:
- Physiological or pathological status
- Congenital physical or mental impairments
- Predisposition to medical conditions or diseases
- Safety/compatibility of treatments
- Treatment response predictions
- Therapeutic measure monitoring
Classic IVD examples include pregnancy tests, blood tests and COVID-19 tests.
IVDs which are approved under the IVDR as suitable for operation in the European market will be granted a CE mark by their EU Notified Body to demonstrate conformity.
How to categorize your in vitro diagnostic device under the IVDR
Like the EU MDR, the IVDR lumps devices into 4 regulatory groups, with low-risk Class A IVDs equivalent to the MDR's Class I medical devices and so on.
Class A IVDs can be self-certified, while all other device classes require assessment and approval by an EU Notified Body.
The IVDR provides 7 'rules' for categorizing devices as follows:
Rule 1: is the device used for detection of high-risk disease in blood, organs, tissues, etc.?
Yes: Class D (highest risk) No: Move onto Rule 2
Rule 2: is the device used for testing blood/ tissue compatibility and whether a high-risk blood group is present?
ABO, Rhesus, Kell, Kidd & Duffy markers: Class D
Immunological compatibility: Class C
No: Move onto Rule 3
Rule 3: is the device used for screening STIs, infectious diseases, cancer, genetics, prenatal, congenital or companion diagnostics?
Yes: Class C No: Move onto Rule 4
Rule 4: is the test a self-test or near-patient test for fertility, pregnancy, cholesterol, or glucose/erythrocyte/leukocyte/bacteria in urine?
Yes: Class B No: Class C
Rule 5: is the test a specific IVD-related reagent, instrument, or specimen receptacle meant for laboratory use?
Yes: Class A (lowest risk) No: Move onto Rule 6
Rule 6: is the device not covered by the previous rules?
Yes: Class B No: Move onto Rule 7
Rule 7: is the device a control used for interpreting other assays, with no qualitative or quantitative value of its own, rather than a diagnostic tool in its own right?
Class B
To learn more about IVDR device classification types and their regulatory pathways, try our classification guide:
When does the IVDR go live?
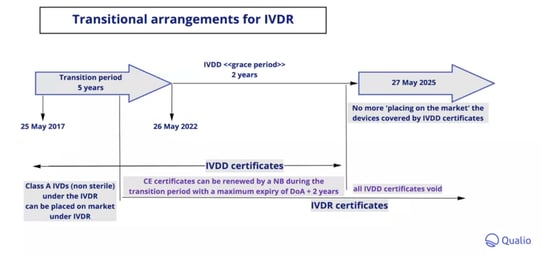
The IVDR originally came into effect on May 25 2017, with a 5-year transition period for organizations to adapt to and meet the new requirements.
From the 'go live' date of May 26 2022, a 'grace period' of 2 years begins where devices can continue to be marketed and sold within the EU with renewed CE certificates issued under the previous IVDD (more on that below!)
These renewed certificates will expire no later than 27 May 2025, at which point all IVDD certificates will be voided and the IVDR will become the sole remaining regulatory category for European in vitro diagnostic devices.
What is the IVDR replacing?
While the EU has had regulation concerning in vitro diagnostic medical devices since 1993, the IVDR's function is to provide a greater level of regulatory detail and scrutiny of how IVDs enter the European markets.
The IVDR repeals Directive 98/79/EC, which came into force in 1998 and became known as the In Vitro Diagnostic Directive or IVDD.
IVDR vs. IVDD: the key differences
Is your business transitioning from the IVDD to the IVDR during the grace period?
Here are the main differences you should know about.
| IVDD | IVDR |
| Classifications based on the conditions the IVD was designed to diagnose; predetermined inflexible list | Classifications based on risk posed by the operation of the IVD. 7 'rules' group IVDs into one of 4 classes, A to D |
| Fairly relaxed classification, with about 93% of devices falling under the lowest risk category, 'IVD Others', and less than 1% under the highest risk 'List A' | Stricter classification: only 16% of IVDs are expected to fall under the terms of the lowest risk Class A. Notified body involvement and regulatory scrutiny in the form of unannounced audits will therefore increase for the vast majority of IVD businesses |
| Limited emphasis on supply chain traceability and post-market surveillance | New clinical evidence standards. Introduction of Unique Device Identification (UDI) system based on FDA practice to strengthen traceability of products. Products to enter public-facing EUDAMED database that collates actors, UDI data, post-market surveillance activity, notified bodies, certificates and clinical investigations and studies |
| Tight definition of IVD: physical devices and receptacles involved in the analysis of bodily specimens | Broader definition to reflect increased digitization: 'software or systems' connected to in vitro activity now fall under IVDR scrutiny like any other device |
For more information about the key changes your business will have to make in the transition to the IVDR, try the EU's May 2022 guidance document, MDCG 2022-6.
IVDR vs. MDR: the key differences
Since the IVDR came into force on the same date as the EU's Medical Device Regulation (MDR) - 26 May 2017 - medical device quality professionals have faced some confusion about the difference between the two standards and any potential overlap.
In short, the IVDR and MDR should be viewed as two halves of a harmonized framework for the governance of medical device quality in the European Union: one half specifically for in vitro devices, the other for all remaining medical device categories.
Let's dive into the main differences.
| MDR | IVDR |
| For non-in vitro medical devices i.e. medical devices which do not have the function of examining/analyzing bodily specimens | In vitro medical device focus |
| Repeals the AIMDD (Active Implantable Medical Device Directive) 90/385/EEC and the Medical Device Directive 93/42/EEC |
Repeals Directive 98/79/EC, the IVDD |
|
Class I (lowest risk) to Class IV (highest risk) device classification |
Class A (lowest risk) to Class D (highest risk) device classification |
|
Pre-market data required:
|
Pre-market data required:
|
|
Post-market clinical follow-up required on a continuous basis |
Post-market surveillance/vigilance required on a continuous basis |
| Notified body involvement required for Class IIa, Class IIb and Class III medical devices | Notified body involvement required for Class B, C and D IVDs |
Key ingredients of IVDR compliance
Your business requires a complete and holistic set of core operational ingredients to make IVDR compliance possible and sustainable.
Let's take a look.
1. Quality management system
An effective quality management system with documented, standardized processes and procedures is vital for IVDR compliance.
Your QMS should encompass all of the following areas:
- Regulatory and compliance strategy
- General safety and performance requirements (GSPR)
- Management responsibility
- Resource management
- Risk management
- Performance evaluation
- Product realization
- Unique Device Identification (UDI)
- Post-market surveillance
- Communication with competent authorities
- Incident reporting & field safety corrective action
- CAPA management
- Monitoring & measurement
Your QMS should be underpinned by strong document and change control.
Ensure the correct references are made within your document stack, consider all the locations where your documents are available, and get a robust process in place for reviewing, approving, updating and re-approving documents as needed.
Annex IX Section 2.4 of the IVDR, meanwhile, specifies that manufacturers need to notify their Notified Body of substantial device or QMS changes - so ensure any changes are properly documented and managed.
Learn more about the core requirements any medical device QMS needs for regulatory compliance
2. Internal auditing
Effective internal auditing will allow you to check and optimize the health and compliance level of your QMS.
For an effective IVDR internal audit program, ensure that:
- The IVDR is documented as an audit criterion
- Your internal auditors are properly trained for IVDR audits (as well as ISO 13485)
- Your audit plan and schedule contains specified and documented IVDR focus
- Your audits, as for any standard or benchmark, are objective and impartial
- All audit activity is documented, audit-trailed and linked to corresponding action workflows
3. Supplier management
Supplier management, encompassing strong control and visibility of your suppliers and critical subcontractors, is crucial now that the IVDR places extra emphasis on the supply chain traceability of IVD products.
Give yourself a mechanism for assessing, onboarding, scoring and communicating with your suppliers in a consistent and audit-trailed way.
In particular, think about:
- Resource management; score the product impact of disrupted supply and get continuity plans in place
- Selection and monitoring, including auditing (see above!) and CAPA plans for any follow-up activity
- Quality agreements and SLAs to bind suppliers and subcontractors to your regulatory requirements
4. Risk management
Risk management is also crucial. As we've seen, the core structure of the IVDR revolves around product usage risk.
A complete and documented risk management strategy is vital if your business is to achieve CE marking for your IVD products.
Your auditor will expect to see:
- Risk management plan
- Risk analysis
- Risk evaluation (as defined in the standard)
- Risk control decision and proposed control measures
- Verification of risk control measures (effectiveness & implementation)
- Assessment of residual risk
- Risk/benefit analysis and report
- Risk management report
- Results of non-conformities, investigations or CAPA activities in manufacturing and/or post-marketing that impact product safety, and any changes resulting from these activities
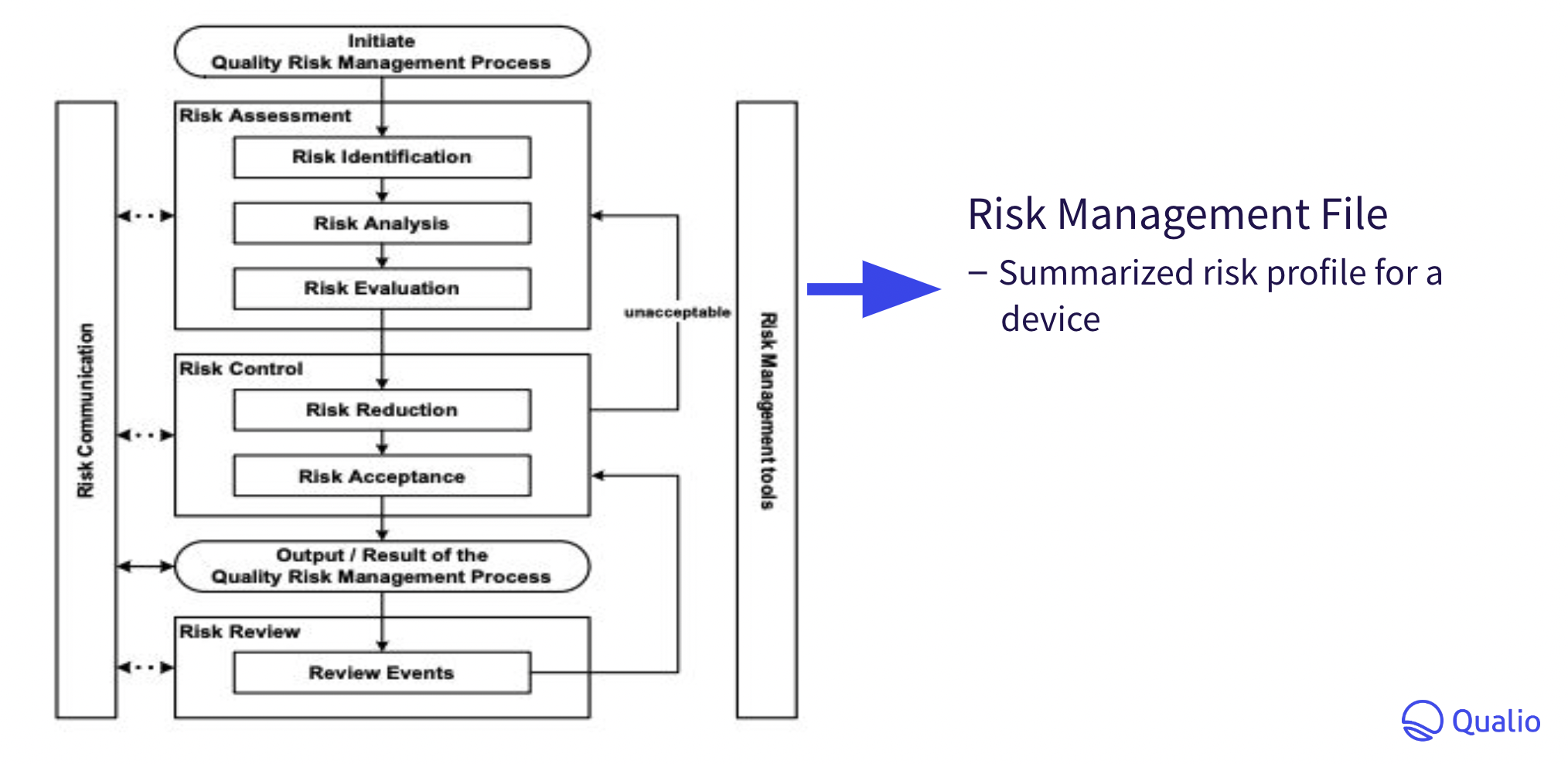
Your risk management file should collate and summarize all risk management activity to generate a risk profile for your in vitro device as follows:
5. Supply chain responsibilities
As we've seen, the IVDR places renewed emphasis on the integrity and traceability of IVD products as they work their way through the supply chain from manufacturer to patient.
For this reason, a harmonized 'pass the baton' relationship with your distributors and importers is just as important as monitoring your own suppliers and ensuring your own quality and compliance as a manufacturer.
For reference, a typical IVD supply chain looks something like this:
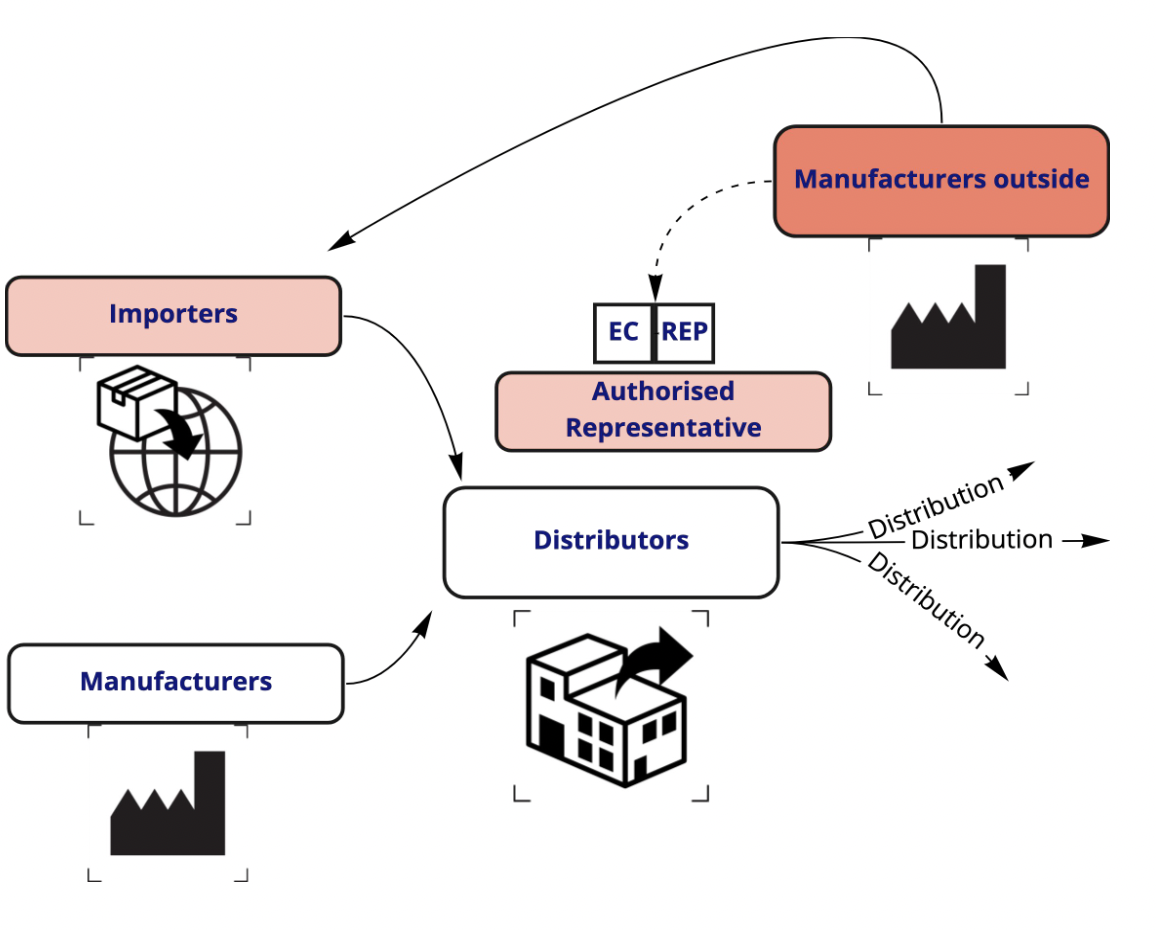
Whether your business is in the EU or not, clear and communicated roles and responsibilities should form an integral part of your import and/or distribution network.
Your distributors and importers both need to:
- Verify your product's CE marking, labelling and declaration of conformity, ensuring only compliant products get to the market
- Provide compliant storage and transportation conditions which don't threaten the general safety and performance requirements of the product
- Cooperate in any corrective actions, including sharing samples and access to product where needed
- Take steps to maximize product traceability
- Inform other cogs in the supply chain, the manufacturer, the Notified Body, or the relevant authorities if any issues arise
6. Post-market surveillance
Your responsibilities as a manufacturer or distributor of in vitro devices don't end on the happy day when your product finally hits the EU's market of half a billion people.
Rather, you should integrate constant post-market performance monitoring into your QMS activity and arm yourself with long-term visibility of the safety and efficacy of your IVD.
Use the intended use and original product statement of your device as the 'Square 1' around which to collect and evaluate post-market evidence.
Above all, your business should form a robust Product Evaluation Plan that includes:
- Development phase outline
- Analytical and clinical performance
- Product development milestones
- Acceptance criteria
- Post-Market Performance Follow-Up (PMPF) planning
GxP is a constantly expanding umbrella. ISO 20916:2019 adds a new middle initial to the set by defining 'Good Study Practice' (or 'GSP') for the completion of clinical performance studies of IVDs, sitting somewhere between the Good Clinical Practice requirements of ICH E6 for pharmaceuticals and ISO 14155 for medical devices.
Top tip: Consider integrating ISO 20916 into your QMS operation to maximize the strength of your post-market surveillance activity
You should also ensure your business can respond to complaints and quality events as they arise. Complaints, for instance, should be responded to within 2 days for critical quality events, or 10-15 days for less serious occurrences.
IVDR compliance tips and tricks
The Qualio quality team has assembled 4 top tips to make your IVDR compliance journey simpler and stronger.
1. Use IVDR terminology
Your QMS document stack should use the same terminology as that outlined in the IVDR, to help your auditor find relevant documentation and to demonstrate your adherence.
2. Look to international standards and guidance for inspiration
We've seen ISO 20916 already. If you haven't already, look to ISO 9001 and ISO 13485 for more detailed quality management and medical device quality management frameworks.
3. Ensure your product file is consistent
Weave a 'golden thread' of clear intended product purpose throughout your technical documentation.
Ensure performance claims are supported with robust scientific evidence, then use your intended device purpose as the benchmark for your clinical evidence capture and evaluation, both pre- and post-market.
4. Consider an electronic quality approach
The IVDR is a highly prescriptive standard that demands a web of controlled and interacting data.
While compliance is possible with manual and paper-based quality systems for very small IVD teams, cost and efficiency ceilings will make long-term compliance difficult as your business grows.
Consider investing in an electronic quality management system (eQMS) to automate and simplify your core IVDR compliance processes, from document and change control to design controls and supplier management.
Learn more
Want more EU IVDR guidance?
Kelly and Sumatha from the Qualio quality team have delivered a comprehensive EU IVDR webinar to help you start your certification journey today.
